
国际罕见病日 | 当罕见病遇见新生儿筛查,一切有了答案
发布时间:2021/2/28 10:56:09 / 浏览量: / 【关闭】
每年二月的最后一天为国际罕见病日,今年是第14个国际罕见病日,今年的主题是“Rare is many. Rare is strong. Rare is proud.”——“罕多,罕强,罕骄傲”。

罕见病日主题故事:6大洲,6张面孔,6位英雄,6条生命。来自全球不同地方不同年龄的6位罕见病患者讲述了他们的故事,Angelina来自澳大利亚,患有X连锁脑桥小脑发育不全伴智力低下和小头畸形(MICPCH)。Tristan 是美国的时装设计师,患有镰状型细胞贫血症。Regina来自巴西,患有平滑肌肉瘤。Syafiq 来自马来亚,患有少汗型外胚层发育不良症(HED)。Harvey来自肯尼亚,患有脊髓性肌萎缩。Jon-Kristian生于挪威,患有成骨不全症。
(一)罕见病并不罕见
罕见病,是一大类发病率很低、大多数具有遗传性的疾病,又称“孤儿病”。事实上罕见病并不罕见,世界卫生组织数据显示,罕见病占人类疾病的10%,约有4亿人饱受罕见病的困扰。中国罕见病患者总数约为1680万,在人群中的综合发病率超过1%。目前已经明确的罕见病有7000多种,其中80%为遗传病。
√新生儿筛查科 是集新生儿遗传代谢病筛查与随访、诊治于一体的专业科室,具备雄厚的技术力量、优良的人才梯队、完善的随访系统和先进的设施、设备。
新生儿筛查科成立于1996年,是青岛市卫健委批准的青岛市唯一一家新生儿先天性遗传代谢性疾病检测与诊治机构,承担全市新生儿筛查、随访与诊治工作。中心拥有先进的串联质谱检测系统、全自动时间分辨荧光检测仪。中心成立伊始即开展先天性甲状腺功能低下(CH)、苯丙酮尿症(PKU)、肾上腺皮质增生症(CAH)和葡萄糖-6-磷酸脱氢酶(G6PD)缺乏四种常规疾病的新生儿筛查。自2011年底始开展串联质谱技术(MSMS)筛查新生儿遗传代谢病,检测范围涉及氨基酸代谢障碍、脂肪酸氧化代谢障碍、有机酸代谢障碍等40余种遗传代谢病的筛查工作,通过筛查,使得部分先天性遗传代谢病可治、可防、可干预,提高其生活质量,同时避免先证家庭同样患儿的出生。新生儿筛查中心将在现有的基础上,继续推进全市遗传代谢病的筛查工作,同时加强与临床科室的联系,对临床上无法明确原因的患儿进行辅助检查,明确其病因,得到早期的诊治。
(二)罕见病与新生儿筛查的“亲密关系”
2018年6月8日,国家卫生健康委员会等五部委联合制定的《第一批罕见病目录》正式发布,涵盖了共121种罕见病,其中有40多种已经有批准上市的治疗药物。我们注意到,这121种罕见病被纳入的条件,与WHO新生儿筛查病种的准入国家标准(Wilson and Junger原则)居然不谋而合:
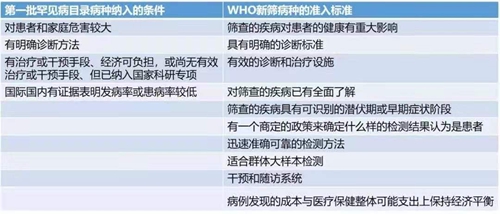
我国最早纳入新生儿筛查的病种是先天性甲状腺功能减低症和苯丙酮尿症,生化和质谱技术的发展又推动先天性肾上腺皮质增生症、葡萄糖-6-磷酸脱氢酶缺乏症、氨基酸、有机酸和脂肪酸类代谢病逐渐纳入新生儿筛查疾病谱。随着分子技术和二代测序的发展,以及治疗遗传病的新药不断涌现,还可以进行糖原累积症、肝豆状核变性、重症联合免疫缺陷病、血友病、耳聋、溶酶体病、杜氏肌营养不良症等疾病的筛查。随着筛查病种的不断增加,更多的罕见病可以在早期发现,并在出现严重症状前给予早期干预,能够很大程度上缩短疾病诊断时间,降低诊断成本,减轻疾病对患者及其家庭身体、精神以及经济上的伤害。
(三)溶酶体贮积症 因治疗药物纳入医保而广受关注
溶酶体贮积症(LSDs),是一组由先天代谢缺陷引起的罕见疾病,疾病种类多达50多种,包括法布里病、戈谢病、糖原贮积病Ⅱ型(又名庞贝氏病)、尼曼-匹克病等,联合发病率约为1/8,000。LSDs会引起周围神经病变,发育迟缓,进行性智力、运动倒退,骨骼异常等,甚至致命,对患者和家庭造成极大的经济和精神负担。然而大部分患者都曾有过误诊的经历,并且往往需要辗转多家医院花费数年时间才能确诊,因而贻误了最佳治疗时机。而通过新生儿筛查实现早期筛查、早期诊断、早期治疗是避免误诊,改善预后,挽救这些患儿的关键。
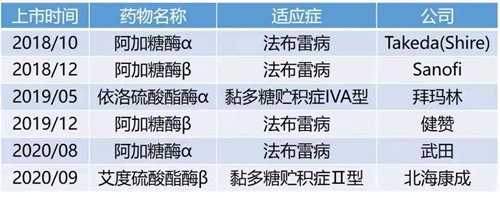
近年来,越来越多的溶酶体贮积症的治疗方案及治疗药物获得突飞猛进的发展。目前已有的LSD治疗方法包括酶替代疗法、造血干细胞移植、底物减少疗法等,其中已获美国FDA或欧洲EMA批准上市的部分LSD治疗药物有近20种;过去的三年,我国也分别审批上市了两种药品用于治疗法布雷病、黏多糖贮积症II型、黏多糖贮积症IVA型,为国内LSD患者带来福音。
由于这些罕见病药物需要长期服用且药价极高,包括全国人大代表在内的社会各方力量竭力奔走呼吁:在全国范围内将戈谢病纳入医保目录!在大家的努力下,LSDs患者终于迎来了曙光:部分自身条件较好的省份首先将戈谢病、庞贝病纳入医保;药企、科研人员加快戈谢病等新药、新疗法的研发;国家也一直在推动通过大病保险、医疗救助、财政专项等方式,解决戈谢病的用药困局。
溶酶体贮积症纳入新筛病种,不再遥远
随着研究的不断深入,干预和治疗方案日趋成熟和规范,美国、意大利的部分地区已经将部分病种纳入常规的新生儿筛查项目,巴西、法国、德国、南非等国家也已经开展试点研究。目前,国内山东等省份也已经开始了溶酶体贮积症新生儿筛查的试点研究。2020年6月20日,由北京健康促进会主办,珀金埃尔默和赛诺菲协办的溶酶体贮积症高危筛查项目正式启动。2020年11月21日中国溶酶体贮积症新生儿筛查协作组成立并开展了2020年国际溶酶体贮积症高峰论坛。社会各方及学术组织的努力,让溶酶体贮积症纳入新生儿筛查已不再遥远。
DMD:钟情于男性的肌营养不良症
DMD——杜氏肌营养不良症,又称假肥大型进行性肌营养不良,为X连锁隐形遗传病,是肌营养不良症中最严重最常见的类型,发病率为1/3000-1/6000活产男婴。DMD患者最初的症状通常出现在3-5岁,包括运动迟缓、步态异常、站立困难及易跌倒等,少数患者还可表现出语言发育迟缓或全面性发育落后。随时间逐渐加重丧失独立活动能力,十几岁时需坐轮椅,并可能出现危及生命的心脏和呼吸系统疾病,通常在20-30岁时死亡。DMD患者的确诊时间一般需要耗费近5年的时间,通过早期的DMD筛查可以缩短确诊时间,改善患者预后,指导患者父母的再生育。
基因治疗,拯救DMD患者的希望
对于DMD的治疗,目前尚未有治愈的手段。临床主要通过药物治疗、物理治疗、基因治疗、以及干细胞治疗的手段对疾病进行控制。近年来,相继有多款新药获批上市,为患者提供了更多的临床用药选择。同时,多种治疗手段均在临床试验各阶段当中,为疾病的治疗和治愈点燃了希望。美国FDA 2020年已获批的53款药品中,13款创新药主要用于治疗罕见病或孤儿病,其中就包括适用于53号外显子跳跃杜氏肌营养不良患者的Viltepso(viltolarsen)。
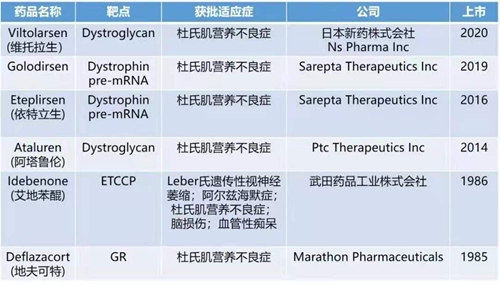
近年全球获批上市的DMD治疗药物
更早推动DMD新生儿筛查的必要性
Annemieke等人的15项有关诊断时间减少和怀疑DMD后续诊断方法的调研内容显示,11项议题中接近100%的家庭强烈认同并推崇,4项内容得到80%以上家庭的认可和推崇,由此可见,缩短DMD确诊时间,推动更早筛查已势在必行。
1975-2011年间,全球多个国家和地区,通过检测干血斑样本中肌酸激酶的水平,完成了180多万新生儿的筛查,发病率为1/4500。美国疾病控制和预防中心也建议,早期诊断可以使DMD患者得到更个性化的治疗。因此,DMD的新生儿筛查也是大势所趋,通过新生儿早期筛查,可以避免误诊,缩短确诊时间,延缓患儿病情发展,有更多的时间等待治疗药物,也可以更好的指导DMD患者父母再生育。
随着政府及社会力量对罕见病防治工作的支持力度不断加大,更多的罕见病能够通过新生儿筛查公共卫生体系达到早筛查、早诊断、早治疗的效果,降低这些疾病对整个人类群体的危害,提高全人类的健康水平和生活质量。